Genomic2DTK - Quick start
Nicolas Chanard
2022-12-08
Source:vignettes/Genomic2DTK.Rmd
Genomic2DTK.RmdQuick start from the import of HiC data to the aggregation of HiC contact in 3D. It includes 4 steps:
- Import HiC
- Import genomic coordinates
- Submatrices extractions
- Plot and visualization
Test dataset
Description
Data were obtained from Drosophila melanogaster S2 cells. HiC test dataset: Directly downloaded from the 4DN platform. Genomic coordinates: ChIPseq peaks of Beaf-32 protein in wild type cells (GSM1278639).
Genomic 3D structure
For a test, please download HiC data in .hic format (Juicer).
options(timeout = 3600)
temp.dir <- file.path(tempdir(), "HIC_DATA")
dir.create(temp.dir)
Hic.url <- "https://4dn-open-data-public.s3.amazonaws.com/fourfront-webprod/wfoutput/7386f953-8da9-47b0-acb2-931cba810544/4DNFIOTPSS3L.hic"
HicOutput.pth <- file.path(temp.dir, "Control_HIC.hic")
download.file(Hic.url, HicOutput.pth, method = 'curl', extra = '-k')Genomic location and annotation data
These kind of data can be imported in R with rtracklayer package.
data("Beaf32_Peaks.gnr")View
| seq | start | end | strand | name | score |
|---|---|---|---|---|---|
| 2L | 35594 | 35725 | * | Beaf32_2 | 76 |
| 2L | 47296 | 47470 | * | Beaf32_3 | 44 |
| 2L | 65770 | 65971 | * | Beaf32_5 | 520 |
Additional genome informations
Required genomic informations used by the functions during the entire pipeline are a data.frame containing chromosomes names and sized and the binSize, corresponding to the HiC matrices at the same resolution.
seqlengths.num <- c('2L'=23513712, '2R'=25286936)
chromSize.dtf <- data.frame(
seqnames = names(seqlengths.num ),
seqlengths = seqlengths.num
)
binSize.num <- 10001 Import HiC
The package supports the import and normalization of HiC data.
Import
Genomic2DTK can import HiC data stored in the main formats: .hic, .cool, .mcool, .h5, .hdf5. The pacakage imports only the raw counts in R. Therefore it is necessary to perform the balancing and observed/expected correction steps.
Balancing
hic.cmx_lst <- BalanceHiC(hic.cmx_lst)
#> No trans matrix, Normalisation won't be applied on trans matricesObserved/Expected Correction
hic.cmx_lst <- OverExpectedHiC(hic.cmx_lst)2 Import genomic coordinates
Genomic coordinates data (ChIP seq peaks or any other feature) need to be indexed using the same referenced genome as for HiC data. Then the genomic coordinates are paired in GInteraction objects.
Features Indexing
Beaf_Index.gnr <- IndexFeatures(
gRange.gnr_lst = list(Beaf = Beaf32_Peaks.gnr),
chromSize.dtf = chromSize.dtf,
binSize.num = binSize.num
)Beaf32 <-> Beaf32 putatives
Constraints for the distance between interaction sites are defined here in order to limit the number of pairs.
Beaf_Pairs.gni <- SearchPairs(
indexAnchor.gnr = Beaf_Index.gnr,
minDist.num = "25KB",
maxDist.num = "100KB"
)3 Submatrices extractions
Once data have been imported, interactions are extracted out of the pairs of genomic coordinates.
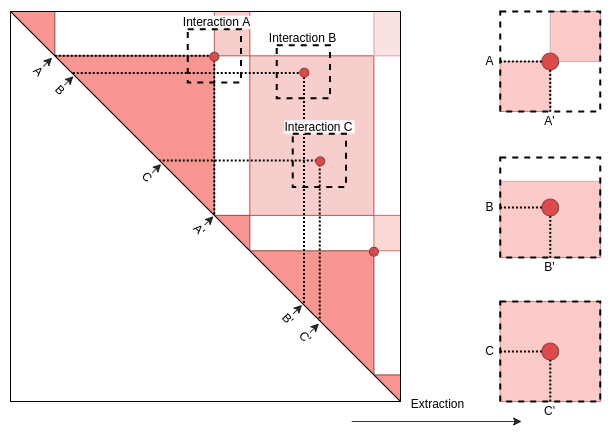
Extraction
Beaf.mtx_lst <- ExtractSubmatrix(
feature.gn = Beaf_Pairs.gni,
hic.cmx_lst = hic.cmx_lst
)4 Plot and visualization
Submatrices are aggregated as sum, average or median. Then, aggregated matrix is plotted as a heatmap of contact frequencies (in the example, contacts surounding Beaf-32 sites).
Aggregation
aggreg.mtx <- Aggregation(Beaf.mtx_lst)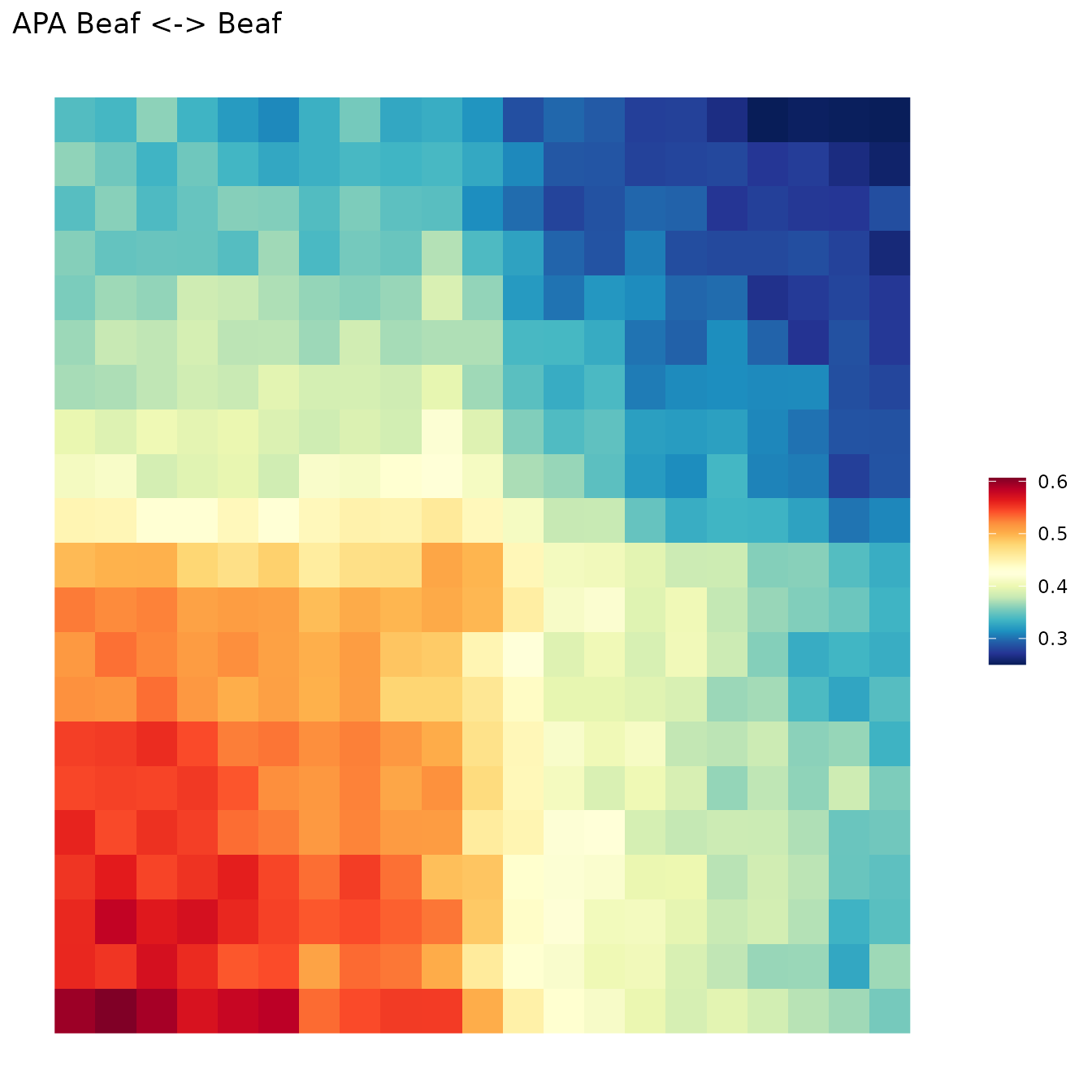
Session Info
sessionInfo()
#> R version 4.2.2 (2022-10-31)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 22.04.1 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] Genomic2DTK_0.99.0 InteractionSet_1.24.0
#> [3] SummarizedExperiment_1.26.1 Biobase_2.56.0
#> [5] MatrixGenerics_1.8.1 matrixStats_0.63.0
#> [7] GenomicRanges_1.48.0 GenomeInfoDb_1.32.4
#> [9] IRanges_2.30.1 S4Vectors_0.34.0
#> [11] BiocGenerics_0.42.0
#>
#> loaded via a namespace (and not attached):
#> [1] Rcpp_1.0.9 lattice_0.20-45 tidyr_1.2.1
#> [4] rprojroot_2.0.3 digest_0.6.30 utf8_1.2.2
#> [7] R6_2.5.1 evaluate_0.18 ggplot2_3.4.0
#> [10] highr_0.9 pillar_1.8.1 zlibbioc_1.42.0
#> [13] rlang_1.0.6 jquerylib_0.1.4 Matrix_1.5-1
#> [16] rmarkdown_2.18 pkgdown_2.0.6 labeling_0.4.2
#> [19] textshaping_0.3.6 desc_1.4.2 BiocParallel_1.30.4
#> [22] stringr_1.5.0 munsell_0.5.0 RCurl_1.98-1.9
#> [25] DelayedArray_0.22.0 compiler_4.2.2 xfun_0.35
#> [28] pkgconfig_2.0.3 systemfonts_1.0.4 strawr_0.0.9
#> [31] htmltools_0.5.4 tidyselect_1.2.0 tibble_3.1.8
#> [34] GenomeInfoDbData_1.2.8 codetools_0.2-18 fansi_1.0.3
#> [37] dplyr_1.0.10 withr_2.5.0 bitops_1.0-7
#> [40] grid_4.2.2 gtable_0.3.1 jsonlite_1.8.4
#> [43] lifecycle_1.0.3 magrittr_2.0.3 scales_1.2.1
#> [46] cli_3.4.1 stringi_1.7.8 cachem_1.0.6
#> [49] farver_2.1.1 XVector_0.36.0 fs_1.5.2
#> [52] bslib_0.4.1 ellipsis_0.3.2 ragg_1.2.4
#> [55] generics_0.1.3 vctrs_0.5.1 tools_4.2.2
#> [58] glue_1.6.2 purrr_0.3.5 parallel_4.2.2
#> [61] fastmap_1.1.0 yaml_2.3.6 colorspace_2.0-3
#> [64] memoise_2.0.1 knitr_1.41 sass_0.4.4